

 ENGLISH
ENGLISH







Introduction
Vitamin D and its hormonally active metabolites are essential for calcium homeostasis and bone health. Growing bones in children become soft and weak in the absence of vitamin D, resulting in a condition known as rickets [1]. The most common aetiology for rickets is vitamin D deficiency due to either poor nutritional sources or inadequate sunlight exposure [2, 3]. In a few rare instances, children do not respond to vitamin D and calcium supplements; this raises the suspicion of either vitamin D-dependent rickets (caused by a 1-hydroxylase or 25-hydroxylase enzyme deficiency) or hereditary vitamin D-resistant rickets (HVDRR; caused by a vitamin D receptor defect).
The HVDRR is a rare autosomal recessive disorder caused by mutations in the vitamin D receptor (VDR) gene. The prevalence of HVDRR is extremely rare, and only around 100 cases are reported in the literature, mostly from regions and countries where interfamily and consanguineous marriages are common (Middle East, Turkey, and Iran) [4–6]. Usually, heterozygous mutations in the VDR gene (in parents of affected children) do not cause signs and symptoms of rickets; however, a few cases have been reported of autosomal dominant mutations resulting in clinical manifestations similar to those of HVDRR [7, 8].
The VDR has 2 terminals: the DNA binding domain (DBD) N-terminal and the ligand binding domain (LBD) C-terminal. The pathophysiology and clinical phenotype of HVDRR depend on which terminal is affected. Mutations in the DBD disrupt the binding of DNA to VDR and cause a loss of gene transcription, while mutations in the LBD result in defects in the ligand (calcitriol) binding [4].
Children with HVDRR present in early life with striking clinical features of rickets, with growth retardation, hypocalcaemia, secondary hyperparathyroidism, hypophosphataemia, and markedly raised 1,25-dihydroxy vitamin D (the active metabolite of vitamin D) [9]. The increase by several folds in 1,25-dihydroxy vitamin D differentiates hereditary vitamin D-resistant rickets from vitamin D-dependent rickets. The clinical picture of HVDRR is unique in that it nearly always presents with partial or total nonscarring alopecia [10]. While the mechanism behind alopecia in HVDRR is not fully understood, it appears that VDR is necessary for hair growth; therefore, when it is affected, inadequate differentiation of pilous follicles occurs, resulting in disintegration and subsequent hair loss [10].
Children with HVDRR are refractory to high doses of vitamin D, to oral calcium supplements, and to supra-physiological doses of active vitamin D analogues (calcitriol and alfacalcidol). Prolonged parenteral calcium infusion therapy promotes mineralisation in bones and normalisation of the biochemical profile [11]. Oral calcium is actively absorbed from the gut via a vitamin D-dependent pathway and is passively absorbed by vitamin D-independent diffusion. High doses of oral calcium supplements (4–6 times the normal daily requirement) facilitate vitamin D-independent diffusion and, in some mild cases, maintain normal serum calcium [12, 13]. Normalisation of hypocalcaemia corrects secondary hyperparathyroidism and thus corrects hypophosphataemia without phosphate supplements [7]. Recently, a few case reports have described the use of cinacalcet (a calcimimetic) as adjunctive therapy in HVDRR, suppressing PTH to improve bone health without causing any instances of severe hypocalcaemia [14, 15].
This case series presents the clinical characteristics, genetic mutations, and treatment with conventional high-dose oral and parenteral calcium in 8 children from 5 Saudi families, as well as our experience with cinacalcet adjunctive therapy in 4 children.
Material and methods
Clinical data
Medical records were reviewed retrospectively to collect clinical data (age at presentation, family history of hereditary rickets, consanguinity, height and weight Z-scores, alopecia, long bone deformities, chest deformities, fractures, and mobility) a biochemical profile (serum calcium, serum phosphate, serum alkaline phosphatase, serum 25-OH vitamin D, and 1,25-OH vitamin D), and renal ultrasound results looking for nephrocalcinosis.
Treatment protocol
Parenteral calcium infusion
The local protocol suggests intravenous elemental calcium (50 mg/kg/day) infusion for 5–7 consecutive days every month in severe cases of HVDRR where oral calcium is not sufficient to maintain a normal serum calcium level until the child completes his or her growth and reaches full puberty. Serum ionised calcium, phosphate, and alkaline phosphatase in addition to renal profile, urinary calcium-creatinine ratio, and parathyroid hormone are monitored during the therapy.
Oral calcium therapy
The recommended dose of elemental calcium in children with nutritional rickets is 30–75 mg/kg/day [16]. We advise taking 4–6 times the recommended dose of calcium carbonate in severe cases to achieve maximum passive absorption from the gut without the action of vitamin D in addition to a monthly intravenous calcium infusion. We adjust the oral calcium dose according to serum calcium and parathyroid levels in mild-moderate cases that do not require intravenous calcium therapy [12, 13].
Cinacalcet
We prescribe adjunctive cinacalcet (calcimimetic) therapy in HVDRR cases when serum calcium levels are within normal ranges with oral and intravenous calcium therapy. Prior to initiation, we obtain consent from parents and we explain the potential risk of hypocalcaemia. The initial dose of cinacalcet used is 0.25 mg/kg/day in 2 divided doses; the dose is titrated to a maximum of 0.5 mg/kg/day based on response [14, 15].
Genetic analysis
We isolated DNA from peripheral leucocytes using the QIAamp DNA Blood Mini Kit (Cat. No. 51104, QIAGEN GmbH, Germany). Using isolated DNA, all exons and exon-intron boundaries of the VDR gene for each patient were amplified by polymerase chain reaction (PCR) using primers and amplification conditions as previously described [17]. The amplicons were verified on a 2% agarose gel and directly sequenced in forward and reverse directions using the dideoxy chain termination method on a 3730XL sequencing machine (Thermo Fisher Scientific, Waltham, Massachusetts, United States).
Results
Eight Saudi children with a clinical diagnosis of HVDRR confirmed with positive VDR gene mutations were treated with either high doses of oral and parenteral calcium or with only high doses of oral calcium; 4 of these children received adjunctive cinacalcet therapy.
Cinacalcet group
Case 1
An 8-year-old girl born to first-degree cousins who first presented at the age of 3 years. Family history was significant for HVDRR in a first-degree cousin. At initial presentation, the height Z-score was –3.2 and the weight Z-score was –2.1; the current height and weight Z-scores were –3.13 and –1.89, respectively. She has normal mobility despite mild bowing of the legs and one long bone fracture; she does not have any chest or spine deformity. Alopecia totalis was present from the initial presentation (Table I). Lab findings at presentation showed hypocalcaemia (2.17 mmol/l), hypophosphataemia (0.83 mmol/l), elevated alkaline phosphatase 1496 U/l, elevated parathyroid hormone (433 ng/l), normal 25-OH vitamin D (60 nmol/l), and elevated 1,25-dihydroxy vitamin D (560 pmol/l) (Table II). She was treated with high-dose oral elemental calcium carbonate (250–400 mg/kg/day). Parenteral calcium therapy was not given because she was able to maintain normal serum calcium and reasonable parathyroid hormone levels with oral calcium alone. Cinacalcet 0.25 mg/kg in 2 divided doses was given for 3 years without any episodes of hypocalcaemia. Median serum calcium was 2.26 mmol/l, PTH was 283 ng/l, and alkaline phosphatase was 951 U/l during the year prior to cinacalcet treatment. During the year with cinacalcet therapy, her median serum calcium was 2.20 mmol/l, PTH was 187 ng/l, and alkaline phosphatase was 1495 U/l (Table III). The renal ultrasound did not detect nephrocalcinosis. Genetic sequencing showed a homozygous truncating mutation of the ligand binding domain at exon 8:c.885C>A:p.Y295X (Table II).
Table I
Patient demographics and clinical characteristics
Table II
Biochemical profiles at initial presentation
Table III
Effect of treatment on serum calcium, alkaline phosphatase, and serum PTH over one year in 4 patients receiving cinacalcet in addition to standard high-dose calcium replacement
Case 2
A 5-year-old boy born to first-degree cousins first presented at the age of 16 months. Family history was negative for HVDRR. He has a history of prematurity and chronic lung disease and is on home ventilation with a tracheostomy in situ. At the initial presentation, his height Z-score was –6.0 and his weight Z-score was –4.3. His current height and weight Z-scores were –6.1 and –6.4, respectively. He is wheelchair-bound with long bone deformities and kyphoscoliosis. He also has alopecia totalis (Table I). The biochemical profile at presentation was serum calcium 2.1 mmol/l, serum phosphate 0.41 mmol/l, alkaline phosphatase 2014 U/l, parathyroid hormone 477.6 ng/l, 25-hydroxy vitamin D 44 nmol/l, and 1,25-dihydroxy vitamin D > 479 pmol/l (Table II). We managed him with monthly parenteral calcium therapy for 5–7 days in addition to high-dose oral elemental calcium carbonate (75–490 mg/kg/day). Additionally, he was treated with cinacalcet (0.3 mg/kg/day) in 2 divided doses for 3 years without any episode of hypocalcaemia. Median serum calcium was 2.22 mmol/l, PTH was 371 ng/l, and alkaline phosphatase was 309.5 U/l during the year prior to cinacalcet treatment. During the year of cinacalcet therapy, the median serum calcium was 2.24 mmol/l, PTH was 199 ng/l, and alkaline phosphatase was 285 U/l (Table III). The renal ultra- sound did not detect nephrocalcinosis. Genetic sequencing showed a homozygous truncating mutation of the ligand binding domain at exon 8:c.885C>A:p.Y295X (Table II).
Cases 3 and 4
Two sisters, aged 9 and 5 years, born to non-consanguineous parents, presented at 23 and 9 months, respectively. Both sisters had alopecia totalis since birth; only the older sister has long bone deformities, and neither of them has a chest or spine deformity. Height Z-scores at presentation were –4.2 and 1.7 while weight Z-scores were –3.5 and –0.9, respectively. Current height Z-scores are 0.6 and –1.58 and current weight Z-scores are 0.36 and –1.21 in the older and younger sisters, respectively (Table I). Biochemical profiles (older and younger sister) were serum calcium (1.68 and 1.92 mmol/l), serum phosphate (0.93 and 1.97 mmol/l), alkaline phosphatase (1,652 and 622 U/l), parathyroid hormone (324 and 213 ng/l), 25-hydroxy vitamin D (5 and 106 nmol/l), and 1,25-dihydroxy vitamin D (479.6 and 381 pmol/l) (Table II). The older sister had a fracture at the first presentation. They both received a high-dose calcium monthly infusion according to the protocol in addition to high-dose oral elemental calcium carbonate (250–400 mg/kg/day). Both sisters were treated with adjunctive cinacalcet (0.3 mg/kg/day) in 2 divided doses; no significant hypocalcaemia occurred while on the treatment. The older sister, one year prior to cinacalcet therapy, had a median serum calcium level of 2.28 mmol/l, a median parathyroid hormone level of 187 ng/l, and a median alkaline phosphatase level of 520.5 U/l. During the year of therapy, her median serum calcium was 2.24, her median parathyroid hormone was 87.5 ng/l, and her median alkaline phosphatase was 378.5 U/l. The younger sister, prior to treatment, had a median serum calcium level of 2.02 mmol/l, a median parathyroid hormone level of 193.3 ng/l, and a median alkaline phosphatase level of 491 µmol/l. While on one year of treatment, her median serum calcium was 2.10 mmol/l, her median parathyroid hormone of 169.3 ng/l, and her median alkaline phosphatase was 428 µmol/l (Table III). Renal ultrasounds were negative for nephrocalcinosis in both sisters. Genetic sequencing showed a homozygous truncating mutation of the ligand-binding domain at exon 6:c.1035 T>C p.Y>X in both sisters (Table II).
Non-Cinacalcet Group
Cases 5, 6, and 7
Three male siblings aged 18, 15, and 9 years born to first-degree consanguineous parents presented at the ages of 36, 24, and 13 months, respectively. They had a first cousin with HVDRR. The middle sibling had the most severe phenotype, presenting with alopecia universalis, a history of bilateral developmental dysplasia of the hips, a history of bilateral humerus fractures, genu varum, chest deformity with kyphosis, limited mobility with limping, reactive airway disease, and the shortest height, with a Z-score of –3.7. The other 2 siblings had alopecia totalis, normal mobility, and no bone deformities; each of them had a final height Z-score of –2.8 (Table I). The biochemical profiles of the 3 siblings at presentation were serum calcium (1.65, 2.37, and 1.89 mmol/l), serum phosphate (0.92, 1.8, and 0.76 mmol/l), alkaline phosphatase (1,409, 313, and 1,407 U/l), parathyroid hormone (362, 252, and 796.4 ng/l), 25-hydroxy vitamin D (10, 63, and 15 nmol/l), and 1,25-dihydroxy vitamin D (> 430, > 432, and 908 pmol/l) (Table II). They were treated with a monthly intravenous calcium infusion according to the protocol in addition to oral elemental calcium carbonate (200–550 mg/kg/day). The parents did not consent to cinacalcet therapy. Genetic sequencing showed a homozygous missense mutation of the DNA binding domain at exon 1:c.88C>T (Table II).
Case 8
A 16-year-old girl born to non-consanguineous parents without any family history of HVDRR presented at nearly 4 years of age with stunted growth, bowing of the legs, and alopecia totalis. She did not have any fractures but developed scoliosis. She was independently mobile with a normal gait. At presentation, her height Z-score was –0.89, her weight Z-score was 0.51, and her current height and weight Z-scores are –2.1 and –0.49, respectively (Table I). Her initial biochemical profile showed a serum calcium level of 2.21 mmol/l, a serum phosphate level of 1.45 mmol/l, an alkaline phosphatase level of 315 U/l, a parathyroid hormone level of 136 ng/l, a 25-hydroxy vitamin D level of 25 nmol/l, and a 1,25-dihydroxy vitamin D level > 479 pmol/l (Table II). She was treated with oral calcium carbonate only; the initial dose was 150 mg/kg/day, and the current dose is 50 mg/kg/day. She did not require an intravenous calcium infusion and her parents did not consent to cinacalcet therapy. She has the mildest phenotype in our cohort. Genetic sequencing showed a homozygous truncating mutation of the ligand binding domain at exon 8:c.885C>A:p.Y295X.
Discussion
This case series describes the clinical presentation, biochemical profiles, genetic mutations, and management of 8 patients with HVDRR. While a few studies have focused on siblings of one family [18] or unrelated patients [4], our sample is unique because it includes 2 sets of siblings (3 brothers and 2 sisters) and 3 unrelated cases. This allows us to observe phenotypic variations in the expression of the same mutations within families.
The clinical characteristics, biochemical findings, and responses to therapy of our patients are comparable to those described in the literature [5] and are delineated in Tables I and II. All 8 of our patients presented with growth failure. Alopecia, a condition commonly associated with HVDRR [5, 19], was a universal finding in our cohort; 7 out of 8 patients presented with alopecia totalis (defined as a complete absence of scalp hair) and one presented with alopecia universalis (defined as a complete absence of all body hair including the scalp, the eyebrows, and the eyelashes). Fractures and pseudofractures frequently occur in HVDRR [1, 4]. In our cohort, 3 out of 8 patients developed at least one fracture; case 6, the most severely affected patient, had multiple fractures in spite of treatment with high-dose oral and parenteral calcium. While most of our patients achieved independent mobility, only one patient out of 8 was wheelchair bound (with co-morbidities including chronic lung disease, a tracheostomy, and being ventilator dependent). It is worthwhile to note that patients who were diagnosed in the first year of life did not develop skeletal signs of severe rickets, which highlights the importance of early diagnosis and intervention. From a biochemical standpoint, elevated alkaline phosphatase, increased PTH, low 25-hydroxyvitamin D, and high 1.25-dihydroxy vitamin D were present in all our cases (Table II). These results are consistent with those previously published [20, 21].
As described by Malloy et al., mutations in the HVDRR gene can be classified into 2 main types: LBD mutations and DBD mutations. The LBD mutations affect the C-terminal end of the gene, which is responsible for binding to vitamin D, while DBD mutations affect the N-terminal end of the gene, which is responsible for binding to DNA [9]. Patients with LBD mutations have a milder phenotype, while those with DBD mutations have a more severe phenotype [9, 10]. The variation in phenotype associated with the type of genetic mutation is probably due to the different ways in which the mutations affect the function of the vitamin D receptor. LBD mutations prevent vitamin D from binding to the receptor, resulting in partial or total unresponsiveness to VDR, while DBD mutations prevent the receptor gene from being transcribed resulting in total unresponsiveness to VDR [19, 22]. These different effects on receptor activity lead to different phenotypes. Similarly, the phenotypic variation among our patients is also consistent with the finding above. For instance, cases 1, 2, and 8 were identified to have the same mutation at exon 8:c.886C>A:p.Y295X, resulting in truncation of the LBD. Consistent with the discoveries of Malloy et al. [4], these individuals exhibited a less severe manifestation of the condition; specifically, cases 1 and 8 were able to sustain near-normal levels of calcium with the exclusive use of oral calcium replacement therapy, indicating a degree of incomplete responsiveness to VDR. Our series also confirms the observation made by Malloy et al. [4] that DBD mutations lead to a more severe phenotype. The genetic investigation of cases 5, 6, and 7 revealed a homozygous missense mutation in exon 1:c.88C>T, which affects the DBD (N-terminal). The patients’ ability to maintain normal calcium levels was solely dependent on regular intravenous calcium administration. Despite treatment, case 6 exhibited significant long bone bowing and kyphosis.
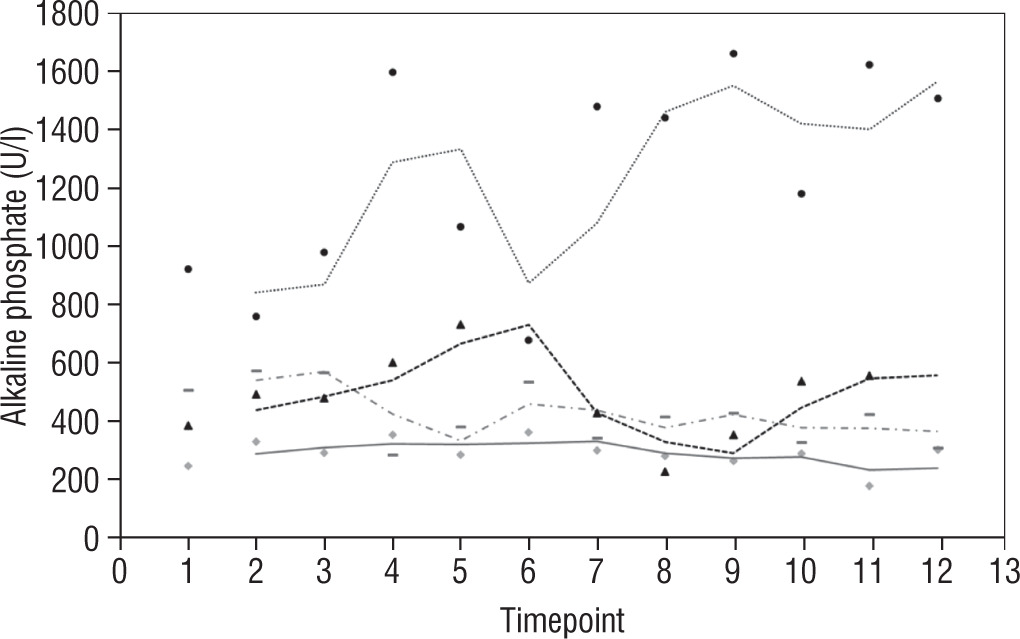
Our study also investigates the safety and efficacy of calcimimetic cinacalcet. Cinacalcet has been used extensively in patients with renal failure, but its use in HVDRR is limited due to concerns that it may cause hypocalcaemia; data in this population are limited to a few case reports recounting the authors’ experience with 1 or 2 patients [14, 15]. In our cohort, cinacalcet was used safely as an adjunctive to high-dose calcium in 4 out of 8 patients; there was no hypocalcaemia following initiation of cinacalcet, nor did this occur at any time throughout the duration of treatment, as seen in Figure 1. When used as an adjunct to standard therapy in HVDRR, cinacalcet is expected to improve PTH levels and other biochemical markers of HVDRR, leading to improved bone health. In our population receiving cinacalcet therapy, following an initial period of suppression, levels of PTH tended to rise (Figure 2); this may suggest that a graded build-up approach to therapy is necessary, periodically increasing cinacalcet doses while monitoring response. Additionally, alkaline phosphatase ALP levels remained unchanged in our cinacalcet group (Figure 3), which is most likely related to the short duration of our study. Further data are required to establish the long-term effect of cinacalcet on ALP levels.
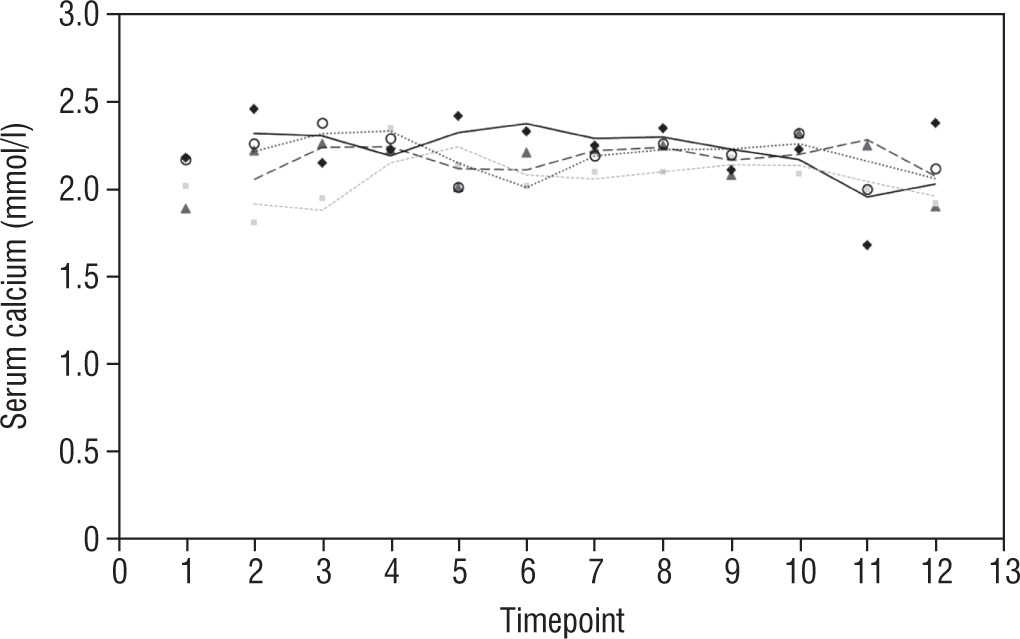
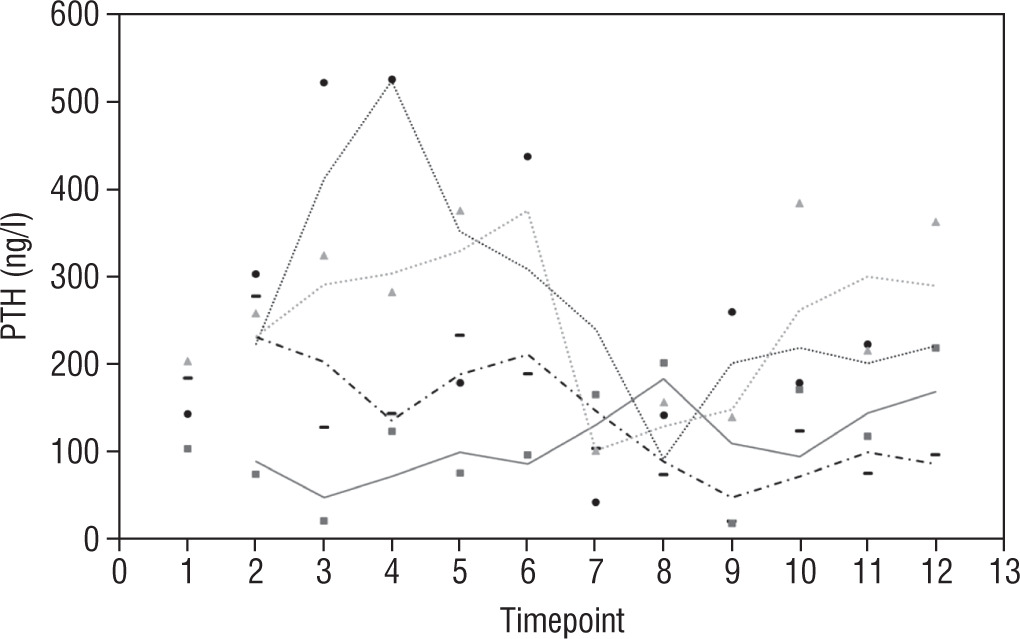
Figure 3
Alkaline phosphatate (U/l) in 4 patients over 2 year period before and after starting treatment
This study has several limitations. First, it is a retrospective study, thereby rendering it susceptible to recall bias. Secondly, the sample size was small and the duration of the study was limited. Despite these limitations, the study provides valuable insights into the clinical features, biochemical findings, and treatment outcomes of HVDRR in Saudi children. The results suggest that cinacalcet is a safe treatment option for children with HVDRR; however, further research over a longer period is required to confirm its efficacy as an adjunctive treatment in this population.
Conclusions
To our knowledge, this is the first case series studying the effect of cinacalcet as an adjunctive therapy to high-dose intravenous calcium in Saudi children, because previous publications consisted of single case studies. While we proved the safety of cinacalcet in patients with HVDRR, structured trials are needed to evaluate the long-term efficacy of adjunctive calcimimetics vs. high dose calcium alone, to improve clinical and biochemical outcomes in this patient population.